Welcome to Computational Mechanistic Chemistry and Drug Discovery
At the heart of our research is a commitment to advancing drug discovery and investigating reaction mechanisms through a variety of cutting-edge methods. We employ techniques ranging from molecular docking to quantum mechanics/molecular mechanics (QM/MM) simulations to explore the processes involved in drug interactions and efficacy. For understanding reaction mechanisms, we utilize straightforward quantum mechanical methods to investigate the underlying principles driving these interactions. Our projects span a broad spectrum, bridging theoretical foundations with practical applications. We focus on method and software development, alongside large-scale molecular modelling that covers everything from small organic compounds to complex biomolecules. This holistic approach allows us to tackle a diverse array of topics, ensuring that our work contributes significantly to the evolving landscape of drug discovery and chemistry.
Our focus areas include:
Library Generation
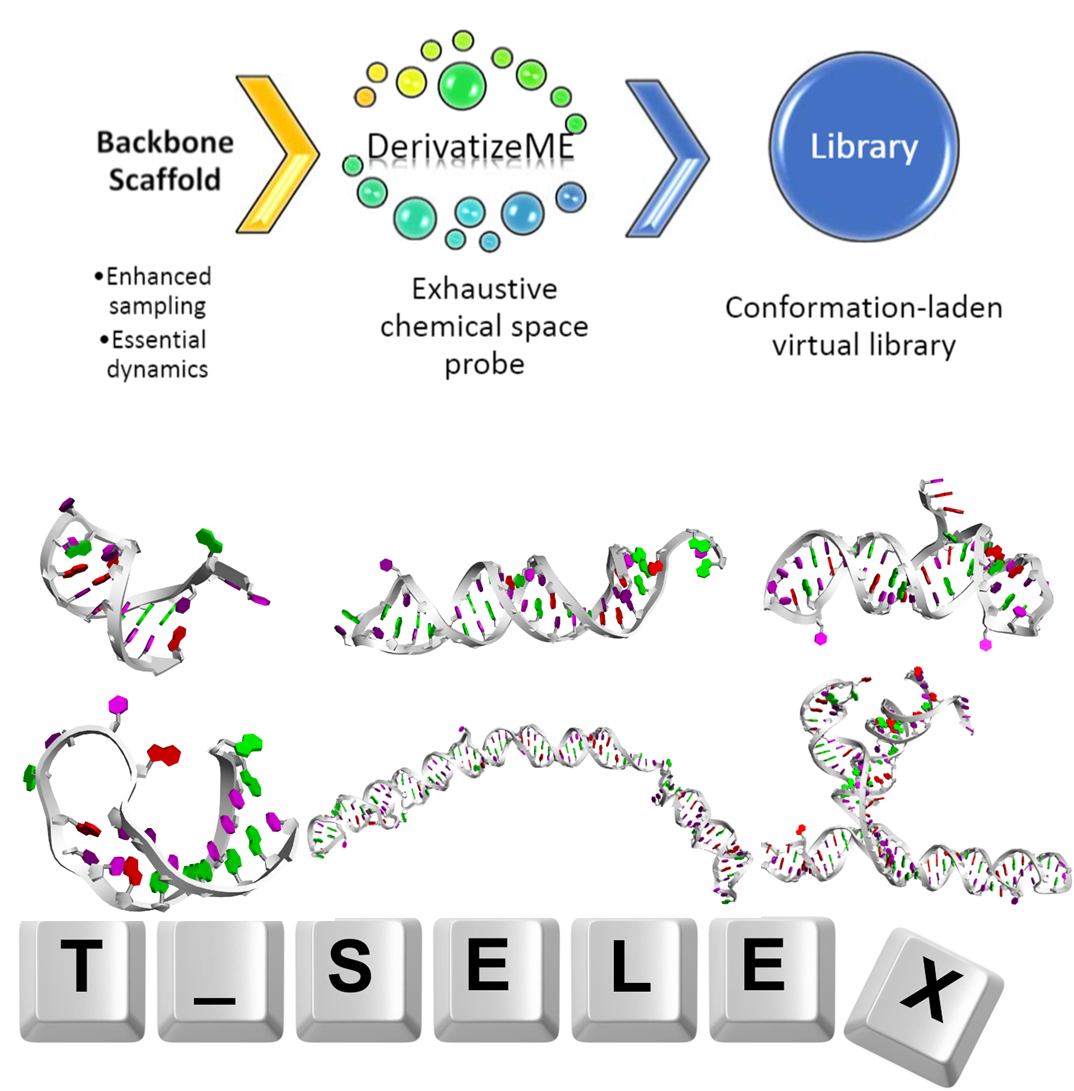
At our research group, we specialize in creating diverse libraries of molecules, both small and large, to explore the vast universe of chemical space. By leveraging natural products as foundational scaffolds, we enhance our ability to discover novel medicinal compounds. Our innovative methodologies combine exhaustive and combinatorial approaches, allowing us to identify compounds with promising medicinal properties for new therapeutic discoveries.
We have developed key programs, including T_SELEX, which enables the large-scale generation and folding of RNA aptamers using the Base Randomization Algorithm. This tool significantly enhances our capacity to create extensive RNA libraries efficiently. Additionally, our DerivatizeME program generates derivatives of specific compounds, transforming those that may not initially meet medicinal criteria into more viable candidates. By shifting these compounds into more promising areas of chemical space, we unlock novel bioactives and privileged scaffolds, driving advancements in drug discovery and development.
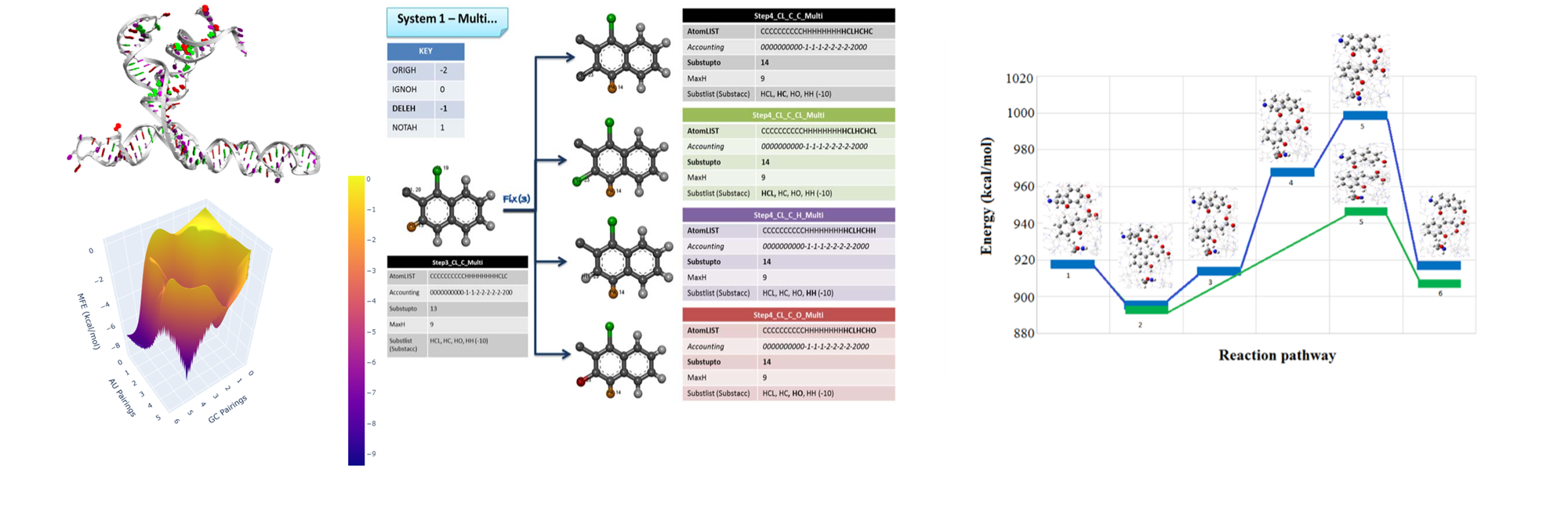
Reaction Mechanism
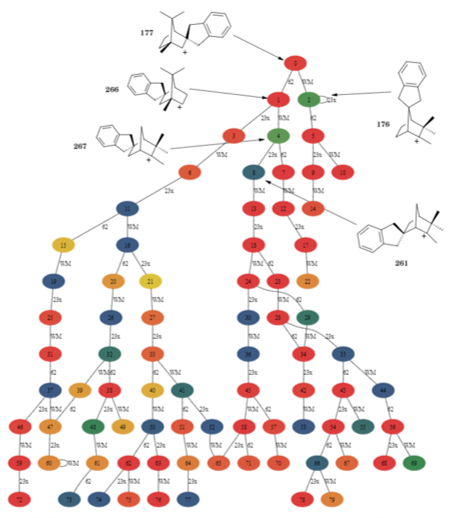
We also investigate reaction pathways to find reaction mechanisms for complex reactions. Our mechanistic approach to chemistry is exemplified by a dedicated program that systematically explores molecular transformations through a Bornane class representing substituted bornane structures. Each structure links to specific descriptors, allowing us to distinguish between variants and enantiomers. We also implement a RuleList class to define preconditions and rearrangement rules for molecular shifts, generating a comprehensive graph of possible transformations. Additionally, we adapted C. K. Johnson's ORNOCARE program for evaluating pathways in norbornyl systems, originally written in PL/1, into C++ using wxWidgets, successfully tested by replicating pathways for acetophenone formation.
Large Scale Reaction Modelling
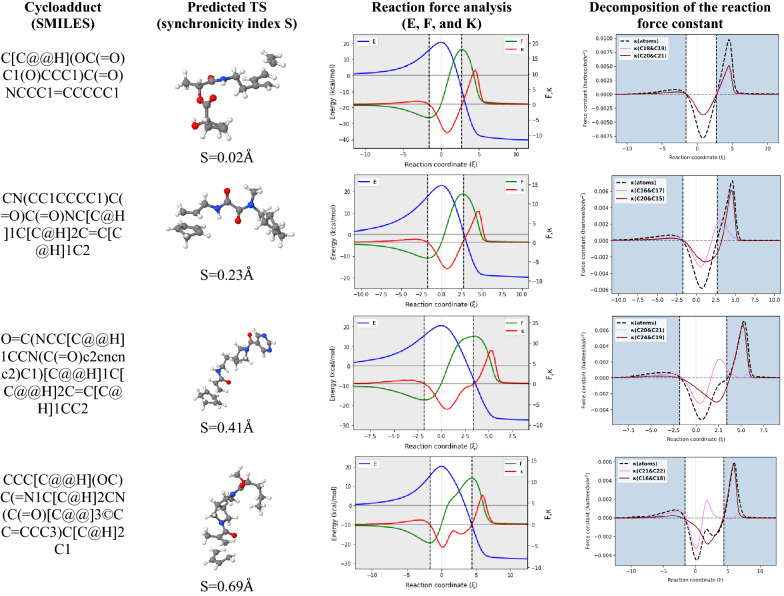
In our research, we also focus on chemical reactions at larger scales. We developed a program called AMADAR, which helps us study reactions in detail and identifies which molecules or substrates can undergo certain reactions. For instance, this program allows us to analyze a wide variety of DA reactions, focusing on their (a)synchronicity and how the reaction proceeds over time. By examining a large dataset of 2,000 potential DA cycloadducts, AMADAR uses methods like reaction force analysis to reveal the mechanisms involved.
Drug Discovery
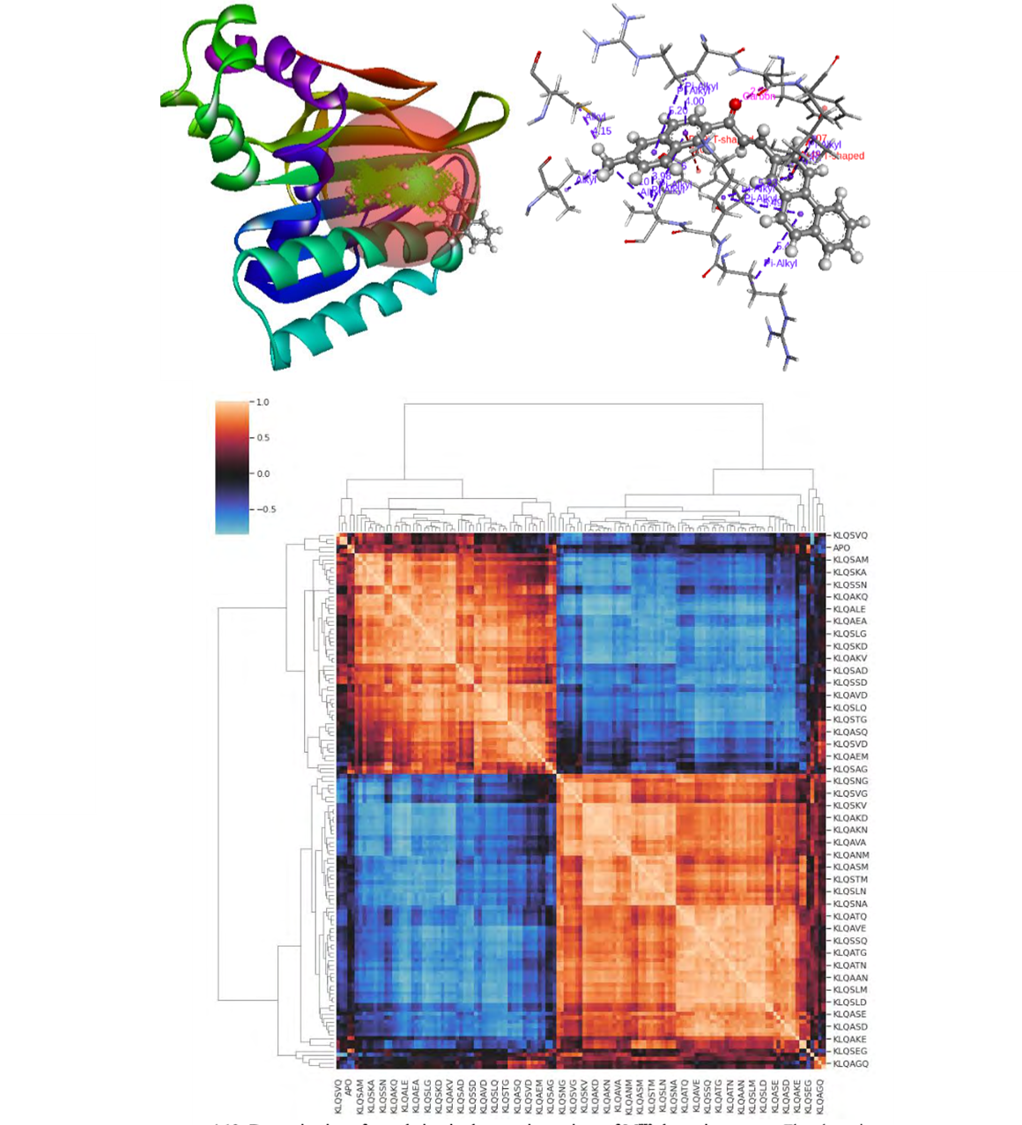
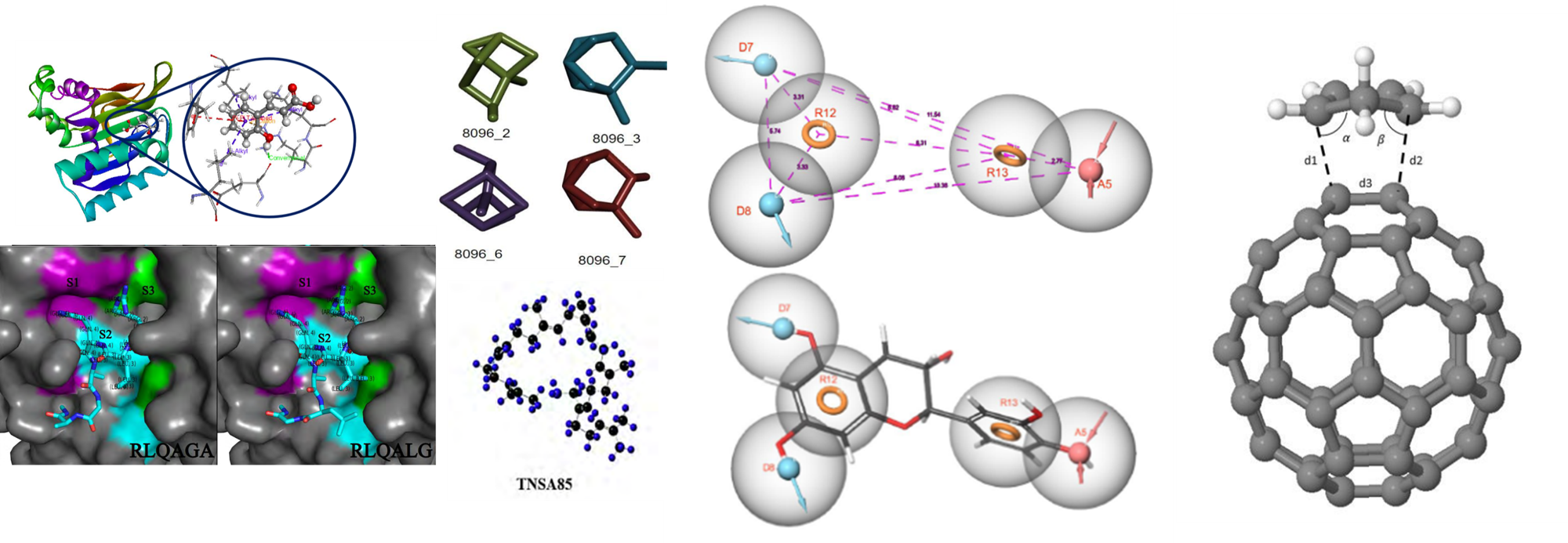
In our ongoing research, we focus on discovering new inhibitors using small molecules, peptides, and aptamers. Our current targets include crucial biosynthesis pathways related to diseases like HIV, SARS-CoV-2, cancer, malaria, and cholesterol metabolism. By exploring these pathways, we aim to identify potential therapeutic agents that could lead to more effective treatments. We look into the enzymatic mechanisms involved in these processes, employing advanced techniques like Quantum Mechanics/Molecular Mechanics (QMMM) to gain deeper understanding.Our ultimate goal is to accelerate the drug discovery process, making it more efficient and effective. We collaborate with experimental chemists and biologists to validate our computational predictions and contribute to the development of groundbreaking therapies.
Machine Learning and Deep Learning
Our research group focuses on the development of advanced artificial intelligence and machine learning methodologies for computational chemistry, molecular modeling, and AI-driven drug discovery. Our research emphasizes the design of novel neural network architectures and graph-based learning systems for molecular representation, reaction mechanism prediction, and quantum-inspired molecular learning. We explore hybrid AI frameworks that integrate molecular graphs, density surfaces, spatial mappings, transformers, reinforcement learning, and physics-informed constraints to enhance chemical prediction accuracy, mechanistic understanding, and model interpretability. A major component of our work involves AI-assisted drug discovery, including molecular property prediction, retrosynthesis, reaction pathway modeling, molecular docking workflows, and adaptive optimization systems for chemical design. In addition, we develop reinforcement learning strategies for ensemble optimization and autonomous scientific decision-making, with the broader goal of creating next-generation intelligent systems capable of learning complex chemical behavior from structural, spatial, temporal, and physicochemical information simultaneously.

Explore our site to learn more about our ongoing projects, team members, and how we are making strides in the field of drug discovery!
Meet Our Team
[Name, Title]: [Brief bio]
[Name, Title]: [Brief bio]
[Name, Title]: [Brief bio]
Our Software Tools
[Software Name]: [Brief description]
[Software Name]: [Brief description]
Who We Are
Founded in [year], we focus on [specific areas of research].
Get in Touch
Email: [contact@example.com]
Phone: [Your phone number]
Join Our Team
[Position Title]: [Brief description]